

Cloacal exstrophy is a very rare condition in which a child is born with their internal abdominal organs — the large intestine and bladder — outside of the body and the bladder is separated into two halves. In boys, the penis is usually flat and short and is sometimes split in two. In girls, the clitoris is separated into two halves.
In addition, some of these organs may not have developed correctly, and the lower parts of the reproductive, urinary and intestinal tracts may not be completely formed. The cloaca is a structure that forms early in development and divides to make the lower intestine and anus, the urinary bladder and the reproductive tract.
Cloaca Development
Normally, the cloaca divides as development progresses. Rarely does this division not proceed in the usual way, and the cloaca remains and may be enclosed within the lower abdomen, or it may be outside the abdominal wall. In the latter case, it’s called a cloacal exstrophy. Structures that may also be affected include the pelvic bones, the spinal column and the spinal cord. It’s a complex birth defect and requires a coordinated care plan developed with input from several specialists to ensure the best outcomes.
Cloacal exstrophy is at the most severe end of a spectrum of malformations called the exstrophy-epispadias complex. Other related conditions on this spectrum include epispadias (rare, but most often seen in boys, a congenital defect in which the urethra opens on the upper surface of the penis) and classic bladder exstrophy (a congenital malformation where the bladder is flattened and exposed on the abdominal wall). Most, if not all, of the conditions on the exstrophy-epispadias complex spectrum require one or more surgeries to repair.
How common is cloacal exstrophy?
Cloacal exstrophy is a rare and complicated condition that occurs during the prenatal development of the lower abdominal wall structures. Cloacal exstrophy occurs in 1 of every 250,000 births. A child with cloacal exstrophy is born with many inner-abdominal structures exposed. A portion of the large intestine lies outside of the body, and on either side of it are the two halves of the bladder. In males, the penis is usually flat and short, and each penile half is separated. In females, the clitoris is also separated into a right half and a left half.
How does it form?
During the formation of organs, while the embryo is in the uterus, the bladder, the urethra, and the front portion of the pelvic bones fail to grow together and fuse in the midline in the front of the embryo. The defect includes the formation of the skin and muscles in the lower abdominal wall. This results in a child being born with an exposed and unclosed on the lower tummy. The inside of the bladder is exposed to the outside air, whereas the back of the bladder is normal.
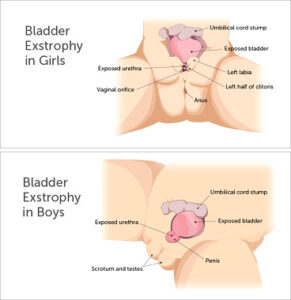
Additionally, in baby boys, the urethra (the urinary tube through the prostate and the penis) is also exposed through the skin and on the top of the penis instead of underneath, known as epispadias. In girls, the urethra is usually shortened and is also split and open to the outside, known as female epispadias; the clitoris is separated into two halves. Because of bladder exstrophy, the open bladder is unable to store urine in any fashion; urine leaks continuously out onto the skin surrounding the affected area
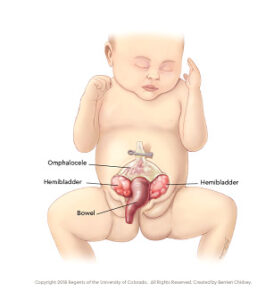
Children born with cloacal exstrophy may also have an imperforate anus, spinal defects such as spina bifida, and/or other organs exposed, including the liver, spleen, colon and rectum (omphalocele).
What causes bladder exstrophy?
The cause is not completely understood but is not caused by anything that the mother does during pregnancy. The defect is believed to occur due to abnormal development of an embryological structure known as the cloacal membrane. This membrane is a temporary structure that allows for the normal development of several pelvic organs and structures, including the bladder, urethra, internal pelvic bones, and muscles, as well as the skin and muscles that cover the structures.
If this membrane persists in the embryo for too long, or possibly if it is overdeveloped, this causes defects associated with bladder exstrophy. As with all other developments, there are always unique aspects to this abnormal development in the individual embryo or fetus with bladder exstrophy. Bladder exstrophy requires surgical repair in order to allow the bladder to store and empty urine.
How is cloacal exstrophy treated?
 The only way to treat cloacal exstrophy is through multiple staged surgeries that:
The only way to treat cloacal exstrophy is through multiple staged surgeries that:
- Aim to separate the digestive tract from the urinary tract, repair the bladder and close up the abdominal wall of the child.
- Closure of the pelvic ring through hip readjustment
- Allow the child the ability to empty urine and stool along with multiple reconstructive surgeries.
Additionally, the child is going to require a lifetime of follow-up care to ensure the proper functioning of the bladder and colon and avoid any chances of kidney failure or bowel obstructions. Furthermore, parents always need to keep in mind that there is no specific blueprint for every child with cloacal exstrophy, and some children are going to require additional surgeries and care to achieve the best possible results.
Common terms
Omphalocele: Some of the abdominal organs protrude through an opening in the abdominal muscles in the area of the umbilical cord. The omphalocele may be small, with only a portion of the intestine protruding outside the abdominal cavity, or large, with many of the abdominal organs (including the intestine, liver and spleen) protruding outside the abdominal cavity.
Exstrophy of the bladder and rectum: The bladder is open and separated into two halves. The rectum and colon are similarly open, and the segment of the rectum is placed between the bladder halves on the surface of the abdomen.
Imperforate anus: The anus has not been formed or perforated and the colon connects to the bladder.
Spinal defects: These defects may either be major or minor. Often children born with cloacal exstrophy are also born with some degree of spina bifida.

 1.405.678.9876
1.405.678.9876